
1. はじめに
「遺伝子を読む」ことは、医療や創薬、祖先解析などを大きく前進させてきました。一方で、全ゲノムを高精度に読むにはコストがかかり、個人レベルで普及させるには壁があります。そこで注目されているのが、“薄く読む(低カバレッジ)”+“統計で補う(インピュテーション)”という考え方です。
この分野では、ハプロタイプ(染色体上の遺伝子の並び)をどう扱うかが鍵になります。ハプロタイプを上手く推定できれば、少ない読み取り量でも、より確からしい遺伝子型やリスク推定につながります。
本記事では、低カバレッジ解析とハプロタイプ推定が、どのように次世代の遺伝子解析コストを下げ、医療応用を現実に近づけているのかを解説します。
図:インピュテーションの基本イメージ(未観測の遺伝子型を参照データから推定)
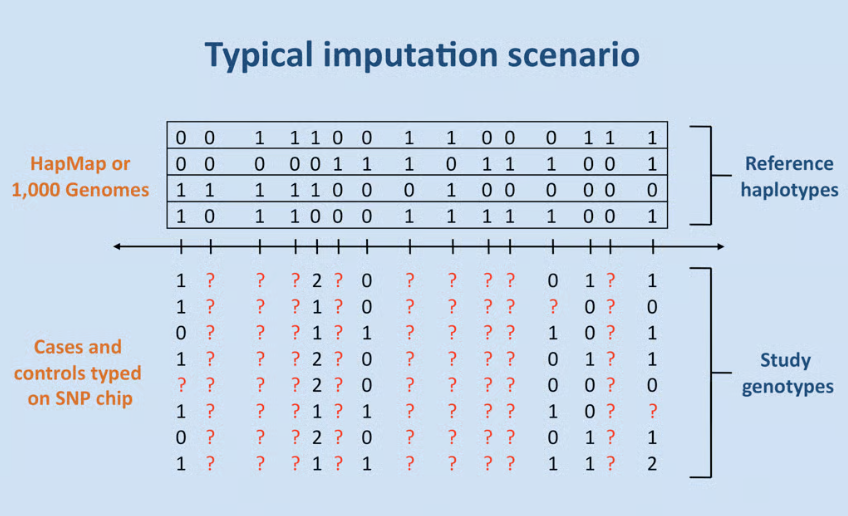
2. 低カバレッジ×ハプロタイプ推定とは何か
2-1. まず「低カバレッジ」とは何か
DNAシーケンスの「カバレッジ」は、同じ場所を何回読むかの指標です。
高カバレッジは確かですが高コストになりやすく、逆に低カバレッジは低コストである反面、読み漏れや不確実さが増えます。
ここで登場するのが、参照パネル(大量の既知データ)を利用し、統計モデルで“もっともらしい遺伝子型”を補うという発想です。これにより、薄いデータからでも、現実に使えるレベルまで精度を持ち上げることが目標になります。
2-2. 「ハプロタイプ」が効いてくる理由
遺伝子型は、SNP(塩基の違い)が点として並ぶ世界ですが、実際には染色体上の並び=ハプロタイプとしてまとまりがあります。
このまとまりをうまく使うと、低カバレッジでも「この人の並びは参照データのこのパターンに近いはず」という推定が効き、欠けた部分を補完しやすくなります。
この仕組みを支える代表的なモデルとして、ハプロタイプを“つぎはぎ(mosaic)”として表現する考え方があり、HMM(隠れマルコフモデル)の枠組みで説明されることが多いです。PLOS Genetics の論文でも、HMMベースのインピュテーションが「観測されていない遺伝子型を推定する土台」になることが図示されています。
図:インピュテーションのシナリオ図(参考パネルから未観測を推定)
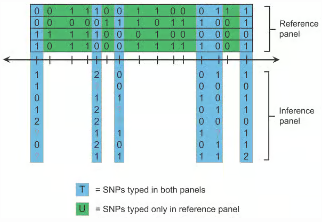
2-3. 「医療・検査」で何が嬉しいのか
この流れが医療に効くポイントは、1人あたりのコストが下がるほど“データが集まりやすくなる”ことです。
たとえば将来的に、妊婦さんの検査(NIPT)や大規模コホートの解析で、低カバレッジを前提にしても十分な精度が出るなら、研究だけでなく臨床データとしても使える範囲が広がります。
実際、低カバレッジの改良を狙う研究として、QUILT2 のプレプリントは 低カバレッジのインピュテーションを高速化し、さらにNIPTのような“複数ハプロタイプが混ざるデータ”にも対応する方向性を示しています。
図:QUILT2モデルの概略(diploid と NIPT モードの考え方)
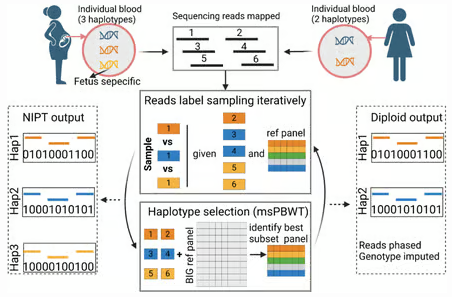
3. 特許から見る技術革新
3-1. 超低カバレッジを“使える”解析にする(US20180044730A1)
この特許は、超低カバレッジ(ultra-low coverage)のシーケンスでも、遺伝子解析に使える形にするという問題意識が見えます。低コスト化を狙う場合、最初にぶつかるのは「読めていない場所が多い」「誤差が目立つ」ことです。
特許の狙いは、薄いデータから得られる情報を整理し、解析・分類・推定へつなげる設計思想にあります。“全部読む”ではなく、“必要な結論に届く読み方”へ最適化する方向性です。
参考:US20180044730A1
3-2. 家族情報×位相推定で「リスク推定」を前に進める(US20150370959A9)
この特許では、家族(クワルテット等)を使った解析の発想が見えます。家族情報があると、単に点(SNP)を当てるだけでなく、“どちらの親由来の並びか”を含めた推定(位相:phasing)が強くなります。
一般的に、位相が分かるほど、リスク推定やHLA領域のような複雑な領域での推定が現実的になります。
参考:US20150370959A9
3-3. 読み取り誤差やサンプル関係推定を“土台”から固める(US20120053845A1)
低カバレッジを現場に持ち込むとき、精度の敵になるのがシーケンス誤差やサンプル間の取り違え・混入です。
この特許はタイトルからも、生体配列の解析とエラー補正、複数サンプルの関係推定を扱うことが分かります。
派手な「新しい診断名」ではなくても、こうした“土台の信頼性”が上がるほど、低コストな解析手法を医療・研究へ載せやすくなります。
参考:US20120053845A1
4. 応用分野・実用化
4-1. 大規模GWAS・コホート(研究の「母数」を増やす)
低カバレッジ+インピュテーションは、高コストな解析を少人数に行うから、ほどほどの精度を大人数に行うへ軸足を移せます。その結果、GWASのように母数が効く研究が進みやすくなります。
4-2. NIPT(出生前)データの高度活用
NIPTは「母体血中のcfDNA」を扱うため、データの性質が一般的な2倍体解析より難しくなります。
QUILT2は、こうしたデータを想定したモード(diploid と nipt)を説明しており、NIPTから母体と胎児の推定に踏み込む方向性を示しています。
4-3. 個別化医療(リスク推定・予防の入り口)
精密医療では、遺伝子型そのものより「どの程度の確からしさで推定できるか」「説明責任を持てるか」が重要です。
特許群が示すのは、精度・速度・コストを同時に満たすための設計(参照データ、位相推定、誤差補正、ワークフロー)が、すでに“実装戦”に入っている点です。
5. 課題と展望
5-1. 現在の課題:参照パネル依存とバイアス
インピュテーションは参照パネルが強いほど精度が上がりやすい一方、参照パネルの偏りがあると、集団によって精度差が出やすい課題があります。
「低コストだからOK」ではなく、どの集団に対しても公平に機能する設計が求められます。
5-2. 研究の最前線:高速化と“実データ形状”への適応
近年の方向性は、単なる精度競争だけでなく、超大規模参照パネルでも回る計算設計、そしてNIPTのような現実のデータ形状(混合・偏り)に合わせたモデルへ進んでいます。QUILT2は、その方向性を明確に述べています。
5-3. 未来の展望:遺伝子解析は「高価な検査」から「基盤インフラ」へ
将来的に、低カバレッジ+推定が当たり前になると、遺伝子解析は“特別な検査”ではなく、医療・創薬・予防の基盤インフラになります。
そのとき重要なのは、派手なAIよりも、誤差を管理し、データの意味を壊さず、臨床で説明可能な形にすることです。特許が示すのは、まさにその実務的な価値です。
あわせて読みたい
同じく「見えない情報を可視化して意思決定を支える」系の技術として、位置推定の発想が分かりやすい回です。
6. 結論
低カバレッジ×ハプロタイプ推定は、遺伝子解析のコスト構造を変える実装技術です。
特許を追うと、「薄いデータをどう使える形にするか」「家族情報や位相推定をどう組み込むか」「誤差補正や関係推定で信頼性をどう担保するか」という、現場で効く工夫が積み上がっています。
高コストで限定的だった遺伝子解析が、より多くの人に届く基盤へ変わっていく——その変化を支えるのが、まさにこの領域の技術革新です。
関連アイテム
ゲノム解析や創薬のニュースで出てくる「バイオインフォマティクス」を、仕組みからやさしく理解したい方へ。
『バイオインフォマティクス入門』で、配列解析・統計・アルゴリズムの全体像を掴む
参考文献
テーマに近い関連する特許文献
- US20180044730A1 – Ultra-low coverage genome sequencing and uses thereof
https://patents.google.com/patent/US20180044730A1/en Source - US20150370959A9 – Phased Whole Genome Genetic Risk In A Family Quartet
https://patents.google.com/patent/US20150370959A9/en Source - US20120053845A1 – Method and system for analysis and error correction of biological sequences and inference of relationship for multiple samples
https://patents.google.com/patent/US20120053845A1/en Source - US20160019341A1 – Methods and systems for genomic analysis
https://patents.google.com/patent/US20160019341A1/en Source
記事を作成するにあたり参考にした文献
- IMPUTE2 公式ドキュメント
https://mathgen.stats.ox.ac.uk/impute/impute_v2.html Source - Howie BN, Donnelly P, Marchini J. A Flexible and Accurate Genotype Imputation Method for the Next Generation of Genome-Wide Association Studies (PLoS Genetics, 2009)
https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1000529 Source - Li et al. Rapid and accurate genotype imputation from low coverage short read, long read, and cell free DNA sequence (bioRxiv)
https://www.biorxiv.org/content/10.1101/2024.07.18.604149v1 Source - Li et al. Rapid and accurate genotype imputation…(full text)
https://www.biorxiv.org/content/10.1101/2024.07.18.604149v1.full-text Source - Fast and accurate genotype imputation in genome-wide association studies through pre-phasing (Nature Genetics)
https://www.nature.com/articles/ng.2354 Source - Flexible read-aware genotype imputation from sequence using …(Nature Communications)
https://www.nature.com/articles/s41467-025-67218-1 Source - Genotype imputation with thousands of genomes(G3, Oxford Academic)
https://academic.oup.com/g3journal/article/1/6/457/5986469 Source
※ 記事は公開されている特許情報および学術研究をもとに作成しています。図版は各出典元から引用しています。
コメント